





Health Economics and Value
Access clearer, more compelling support to demonstrate the value of your product.



Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more

Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more
Developing IQVIA’s positions on key trends in the pharma and life sciences industries, with a focus on EMEA.
Learn more
"We strive to help improve outcomes and create a healthier, more sustainable world for people everywhere.
LEARN MORE
Reimagine clinical development by intelligently connecting data, technology, and analytics to optimize your trials. The result? Faster decision making and reduced risk so you can deliver life-changing therapies faster.
Research & Development Overview
Generate and disseminate evidence that answers crucial clinical, regulatory and commercial questions, enabling you to drive smarter decisions and meet your stakeholder needs with confidence.
REAL WORLD EVIDENCE OVERVIEW
Elevate commercial models with precision and speed using AI-driven analytics and technology that illuminate hidden insights in data.
COMMERCIALIZATION OVERVIEW
Orchestrate your success across the complete compliance lifecycle with best-in-class services and solutions for safety, regulatory, quality and medical information.
COMPLIANCE OVERVIEW
When your destination is a healthier world, making intelligent connections between data, technology, and services is your roadmap.
TECHNOLOGIES OVERVIEWExplore our library of insights, thought leadership, and the latest topics & trends in healthcare.
DISCOVER INSIGHTSAn in-depth exploration of the global healthcare ecosystem with timely research, insightful analysis, and scientific expertise.
SEE LATEST REPORTS
By making intelligent connections between your needs, our capabilities, and the healthcare ecosystem, we can help you be more agile, accelerate results, and improve patient outcomes.
LEARN MORE
Building on a rich history of developing AI for healthcare, IQVIA AI connects the right data, technology, and expertise to address the unique needs of healthcare. It's what we call Healthcare-grade AI.
LEARN MORE
The IQVIA Human Data Science Cloud is our unique capability designed to enable healthcare-grade analytics, tools, and data management solutions to deliver fit-for-purpose global data at scale.
LEARN MORE
The IQVIA Innovation Hub connects start-ups with the extensive IQVIA network of assets, resources, clients, and partners. Together, we can help lead the future of healthcare with the extensive IQVIA network of assets, resources, clients, and partners.
LEARN MORE
IQVIA Decentralized Trials deliver purpose-built clinical services and technologies that engage the right patients wherever they are. Our hybrid and fully virtual solutions have been used more than any others.
LEARN MORE
Empowering patients to personalize their healthcare and connecting them to caregivers has the potential to change the care delivery paradigm.
LEARN MOREOur mission is to accelerate innovation for a healthier world. Together, we can solve customer challenges and improve patient lives.
LEARN MORECareers, culture and everything in between. Find out what’s going on right here, right now.
LEARN MORE
"Improving human health requires brave thinkers who are willing to explore new ideas and build on successes. Unleash your potential with us.
SEARCH JOBSWe are at the dawn of a new era for European Health Technology Assessment (HTA) and the recent publication of the draft Joint Clinical Assessment (JCA) for medicinal products Implementing Act (IA) brought us one step closer. The purpose of this much anticipated Act is to provide more details on how the EU HTA process will function in accordance with the HTA Regulation (HTAR). As IQVIA experts outlined in the white paper “The future of EU HTA”, previous guidance developed by EUnetHTA 21 left many questions unanswered regarding timelines, the scoping process and stakeholder involvement, as well as what the process would look like in practice. This blog explores what has changed in the IA on JCA, what has not been addressed, and how to prepare for 2025 and beyond.
Background on the Implementing Act: Six IAs are outlined in the HTAR, with the draft of the IA on JCA for medicinal products the first to be published. Initially expected in December 2023, long debate and multiple meetings by the European Commission (EC) committee on HTA delayed the publication until March 5th ,2024.
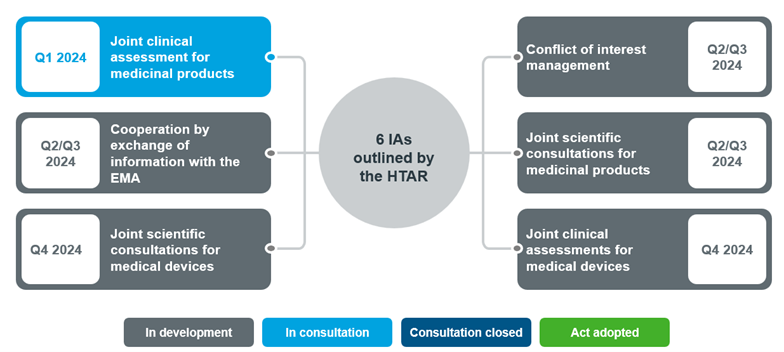
Fig 1. Overview of Joint Clinical Assessment/ Joint Scientific Consultations Implementing Acts expected in 2024
Source: Implementation Rolling Plan February 2024.
Note: These are the anticipated timelines presented in February 2024
Following EC comitology procedure, the public can now provide feedback on the draft until April 2nd, 2024 after which the IA will be adopted. The other IAs will follow before the regulation enters into force from January 12th, 20251.
This draft IA provides guidance on the start of the JCA process, interaction with stakeholders, selection of experts, scoping, dossier submission and JCA production including format and templates. It builds on the outcomes and documents produced by EUnetHTA joint actions and the more recent EUnetHTA 21 service contract. However, the IA only sets out the procedural rules; further details on methods and processes are expected in guidance documents, which are currently being developed by the Methodological and Procedural Guidance (MPG) Subgroup.
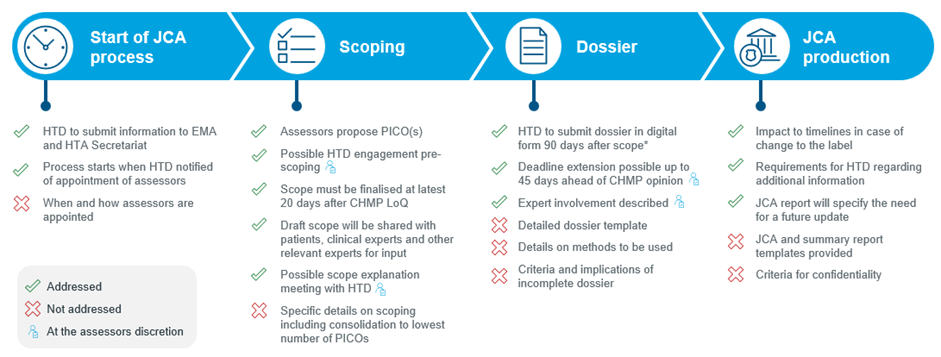
Fig 2. Overview of the draft JCA Implementing Act
*60 days for accelerated procedures or type II variations
Start of JCA process: We see a bit more involvement of the health technology developer (HTD) in the JCA process. The HTD is required to submit the proposed summary of product characteristics (SmPC) and clinical overview section of the EMA submission file to the HTA secretariat at the same time as the EMA submission, which is used by the assessors to inform the population, intervention, comparator and outcome (PICO) proposal. The IA also specifies that EMA will notify the HTA secretariat when it receives the submission. However, the HTD is not notified of any communications between the HTA secretariat and EMA at any point in the JCA process.
Expert consultation: The role of experts and stakeholders has been elevated, with increased and more formalized involvement throughout the JCA process. Selection of experts will be conducted by the JCA subgroup, with the HTA Secretariat expected to provide a list of potential candidates. Priority will be given to experts with experience covering multiple member states (MS). The importance of expert engagement is highlighted, which allows the assessors to consult experts during the drafting and finalization of the assessment scope and drafting of the JCA and summary report.
Scoping: Timelines for scoping have been confirmed, some potential additional input from the HTD have been introduced, and the changes to the survey process that were included in the updated EUnetHTA 21 Scoping Guidance are confirmed. Following receipt of information from the HTD for scoping, the JCA subgroup may invite the HTD to provide further input in a meeting with the JCA subgroup or in writing. As indicated in the EUnetHTA 21 Scoping Guidance, the assessors will draft a proposed PICO based on the information provided by the HTD and will share this with member states (MS) via a survey. The exact starting date of the scoping process is not specified, but the JCA subgroup will finalize the assessment scope at the latest 20 days after the Committee for Medicinal Products for Human Use (CHMP) adopts its list of questions, or 140 days after submission of the regulatory file. There is a possibility for the HTD to be invited to a scope explanation meeting with the JCA Subgroup within 30 days after the JCA scope has been shared. Whether this meeting takes place is at the discretion of the JCA subgroup. It is understood that the scope explanation meeting will be reserved for very complex JCAs and will not be standard for every submission.
Dossier submission: Timelines for the dossier submission have been confirmed. For the standard EMA process, HTDs will need to submit the dossier in digital format 90 days after they have received the final scope. In cases of an accelerated regulatory procedure or extension of indication, HTDs will have only 60 days to submit the dossier. There is some flexibility with the possibility of an extension to the deadline in ‘justified cases’. However, this is at discretion of the assessors and is anticipated only in exceptional cases. The dossier will need to be submitted at the latest 45 days prior to the envisioned CHMP opinion, as laid out in the HTAR.
A dossier template outline was provided in Annex 1; this is largely consistent with the template provided by EUnetHTA 21, with some new elements related to justification for the omission of PICOs, patient registries, and availability of new data relevant for the scope. More detailed guidance on the template is expected to be published later this year. The draft IA stipulates that in situations which the HTD does not provide evidence for a requested PICO, a rationale should be provided. HTDs will now be required to include searches of patient registries in addition to study registries. This aligns with the increased involvement of experts including patient organisations. Finally, HTDs must now report whether and when new data relevant to the scope that might lead to an update of the JCA might become available.
JCA production: A template, similar to the EUnetHTA 21 draft, has been provided for the development of the JCA and summary report. Some indication on the implication of a label change was also provided. If this situation arises, the JCA Subgroup will decide if JCA will proceed and will notify the HTD. If the label change does not result in a change to the JCA scope, then the JCA report will be published in 180 days from re-initiation. This is increased to 330 days if the label change results in a change to the JCA scope.
The timelines for responding to requests from assessors for additional information/analyses is notably very short: 15 days (10 days if an accelerated procedure/ type II variation). In addition, at any time during the development of the draft JCA, the assessors may ask the HTD for additional information, data, analyses or other evidence, with a deadline of between 7- 30 days. The HTD may also submit new clinical data up to 7 days after the adoption of the CHMP final opinion.
The JCA report may indicate the need for an update (e.g., when new data becomes available), in which case it is the HTD’s responsibility to inform the Coordination Group when the new data will be available. The update will be included in the group’s annual work program and where possible, the same assessors as the original JCA will be assigned.
Start of the JCA process: The JCA process starts once the HTD is notified of the appointment of the JCA assessor/ co-assessor. It is not clear exactly when this will be, but this is likely in line with EUnetHTA 21’s original proposal, which means the scoping process will kick off 1-2 months after EMA submission. In case of a label change during the process, timelines for completion the JCA are specified; however it is not known how long it will take to assess the implications of the label change and when this will be communicated to the HTD.
Scoping: Most industry requests were left unanswered, with the scoping process to a large extent remaining a black box, albeit the Methodological and Procedural Guidance (MPG) Subgroup is currently conducting further PICO exercises and is expected to publish their PICO guidance later this year. Whilst the HTD now must provide information to inform the scope, which will give an opportunity to provide input into the scoping proposal, it is not clear how this and other inputs will be used by the assessor to develop their PICO proposal. In addition, beyond the scope proposed by the assessors, it is unclear what additional information will be provided to MS alongside the proposed PICOs. The IA specifies that the JCA subgroup will discuss the scope during a consolidation meeting, but as no expectations are set in terms of number of PICOs, the risk of a high number of PICOs in the final scope remains.
Dossier: The template provided in the IA annex leaves many unanswered questions and concerns about when the “table collection” will be made available. Regarding methodology to be used, the template instructs HTDs to follow methodological guidance adopted by the HTACG. The MPG subgroup is developing guidance on direct and indirect comparisons, validity of clinical studies, and endpoints, but none of these are available as of March 12th, 2024. The template also includes a section in which HTDs should provide a justification for not providing results for requested PICOs, however, no clarity was provided as to what would be an acceptable justification for these instances and what would be considered an incomplete dossier.
JCA production: The largest concern for HTDs regarding the JCA report is related to confidentiality of the information presented. HTDs will have 7 days after they receive the draft JCA and summary report to check factual and technical accuracy and identify confidential information and will be required to justify its commercial sensitivity. The European Commission (EC) will take the JCA subgroup viewpoint on HTD requests for commercially confidential information, however there is no certainty that requests will be accepted and there is a risk that commercially sensitive information could be published. Moreover, the HTD is asked to check the revised draft JCA but will not have the opportunity to see any input from other stakeholders before it is finalised by the JCA subgroup, unless the HTD is invited to the meeting to discuss the input on the revised draft JCA report.
The much-anticipated draft JCA IA is undoubtedly a step towards the transparency that industry has been seeking; however, we must remember that this is just the first draft IA, which provides high-level procedural guidance. Comments may be addressed in the final IA, although there is no requirement to do so. Nevertheless, keep watching this space as there is more to come from the MPG subgroup who will publish further guidance on methodology later this year.
Now that we have clarity on timelines, the JCA process needs to be planned with military precision. Aside from bringing global and local interactions forward to anticipate PICO requests, very close collaboration with regulatory teams is also required. The dossier will need to be initiated 4 months ahead of EMA filing and it is recommended to draft the full dossier in advance of receiving the final scope. Once the scope is confirmed, the timelines for JCA dossier submission are very short and overlap with the response to the EMA `Day 120´questions. The timelines are even shorter for an accelerated EMA process, and therefore a careful benefit-risk assessment of an accelerated process will be required. Post-submission, there is an extremely short turn-around time for responding to data requests and the factual accuracy check, therefore careful consideration will need to be given to internal resource allocation for the EMA/ JCA processes.
It is unlikely that evidence requirements will be relaxed and therefore a meticulous cross-functional JCA strategy will need to be developed and executed, considering all possible PICO scenarios and evidence requirements. As we await the publication of the MPG methods guidance, we anticipate that points of view on topics, such as the use of single arm trials, will be align with those included in the EUnetHTA 21 methodological guidance. Integrated evidence generation plans will need to incorporate JCA requirements, the systematic literature review (SLR) protocol will need to be carefully considered so as not to exclude relevant comparators, and a refresh will need to be planned three months prior to the JCA submission. Whilst the door is opened for omitting some PICOs, a thorough indirect treatment comparison (ITC) feasibility will need to be carried out to ensure justification is accepted. If it is not accepted, there is only two weeks to provide the missing information.
The impact on local submissions should also not be forgotten, as any PICOs not requested by JCA will most likely subsequently be requested at a local level. As the JCA process evolves, so will processes at a MS level. Monitoring local implementation and adoption i.e., changes to evidence requirement or dossier templates, will be key to ensuring seamless transition from JCA to patient access at a MS level.
HTD should capitalize on opportunities to engage with stakeholders to influence the JCA scope. Although there is no formal letter of intent, it may be possible to make a case for relevant PICOs, when submitting information to the HTA secretariat. We now see stakeholder engagement throughout all steps of the JCA process from scoping to review of the draft JCA. Early local stakeholder engagement will also be key to seek input on and shape JCA strategy.
Finally, HTDs should carefully consider the justification of commercially confidential information in the draft JCA, as there is a risk that without clear justification, commercially sensitive information may be published. As the HTD is asked to review the revised draft JCA, it is advised to also check the final published JCA for accuracy.
Considering all of this, the road to JCA adoption may seem long and uncertain. Carefully considered early cross-functional planning and preparation will lay the foundation for a successful JCA and aid a smooth transition into this new process.
More insights on EU HTA be found on IQVIA's dedicated EU HTA page. Learn more about topics such as "How emerging biopharma companies can prepare for EU HTA" and EU HTA for medical devices. More information will be published as we approach 2025.
1 Implementing and delegated acts - European Commission (europa.eu)
Access clearer, more compelling support to demonstrate the value of your product.
Get insights into payer decision-making and evidence requirements to accelerate market access for your drug.